La rete delle conoscenze nefrologiche
Attenzione! Per visualizzare al meglio il sito e usufruire di tutte le funzionalità messe a disposizione
si consiglia di aggiornare la versione in uso di Internet Explorer alla versione 8 o superiore. Grazie!
La rete delle conoscenze nefrologiche

Home > Articoli originali
Pubblicato il 10 novembre 2016
Scarica l’APP del GIN per il tuo smartphone o tablet:


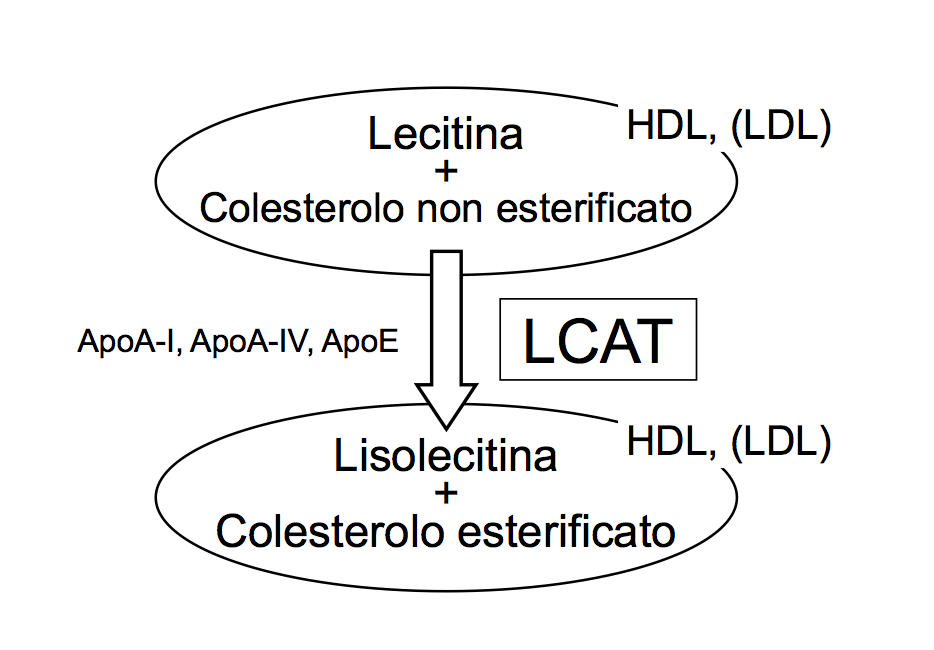
Fabio Lucca1, Alice Ossoli1, Giuliano Boscutti2, Guido Franceschini1, Laura Calabresi1
(1) Centro E. Grossi Paoletti, Dipartimento di Scienze Farmacologiche e Biomolecolari, Università degli Studi di Milano
(2) Nefrologia e Dialisi, Azienda Sanitaria Universitaria Integrata di Trieste
Corrispondenza a: Laura Calabresi; Centro E. Grossi Paoletti Dipartimento di Scienze Farmacologiche e Biomoleculari Università di Milano Via Balzaretti 9 20133 Milano Italy; Tel: +39 0250319906; Fax: +39 0250319900; E-mail: laura.calabresi@unimi.it
© 2013-2025 Società Italiana di Nefrologia — ISSN 1724-5990 — Editore Tesi SpA
Giornale Italiano di Nefrologia è una testata giornalistica registrata presso il Tribunale di Milano. Autorizzazione n. 396 del 10.12.2013.
La piattaforma web su cui condividere in maniera semplice, efficace ed interattiva le conoscenze nefrologiche attraverso la pubblicazione online di documenti multimediali.
NephroMEET accoglie come documenti con marchio SIN quelli approvati da: Comitati e Commissioni ufficiali SIN, Gruppi di Studio SIN, Sezioni Regionali/Interregionali SIN.
Il Consiglio Direttivo SIN si riserva inoltre la facoltà di certificare con marchio SIN altri documenti qualora lo ritenga opportuno.
Gli Autori si assumono in ogni caso la responsabilità dei contenuti pubblicati.
I contenuti pubblicati sono riservati ad un pubblico esperto nel settore medico-scientifico.
Seguici su Twitter
Developer e partner tecnologico:
TESISQUARE®
Assistenza telefonica allo 0172 476301
o via mail